Definition of data types (as used in ENA)
-
‘Raw’ sequence data - Sequence data that is obtained directly from a sequencing instrument (e.g. in FASTA, FASTQ, BAM, or CRAM files). ENA recommends the use of FASTA/FASTQ file formats (described below), but accepts also other file formats.
-
Analysed sequence data - Sequence data that has been processed in some way after being obtained from a sequencing instrument. Such data has been normalised, and perhaps also subject to other processing (e.g. removal of outliers, calculation of expression measurements, and statistical analyses).
-
Metadata - Description of the data that gives, at a minimum, sufficient information to reproduce the data collection method (e.g. description of how the source material was obtained and details about the sequencing process, such as library preparation and the instruments used). In ENA, all metadata related to a research project is represented by different types of metadata objects. See below for an explanation of different types of objects.
Definition of metadata objects
ENA recognises multiple ’levels’/’types’ of metadata related to sequencing projects. These different ’levels’/’types’ of metadata are represented by different metadata objects. For example, general project information (such as the project title) is defined in the study (project) object, sequenced source material is included in the sample object, and details about the sequencing experiment are captured in the experiment object.
Below is more information on what each type of metadata object comprises:
-
Study - A study (project) object is used to group together all data submitted to ENA about a given study and to control its release date. A study accession number is typically used to cite data submitted to ENA. Note that all data and metadata associated with a study are made public together with the study when it is released.
-
Sample - A sample object contains information about the sequenced source material. Checklists are in place to define which fields should be filled when annotating samples. Note that a taxonomic classification system is used to refer to biological organisms; the accepted organism name and classification hierarchy are used, see ENA taxonomy for further details.
-
Experiment - An experiment object contains all the details about the metholodology used for sequencing, including library and instrument details.
-
Run - A run object is part of an experiment object. It refers to data files that contain ‘raw’ sequence reads.
-
Analysis - An analysis object contains secondary analysis results. These results are derived from ‘raw’ sequence reads (e.g. a genome assembly).
The different metadata objects relate to different stages of the sequencing process. A summary of which metadata objects relate to which stages is shown in the below figure.
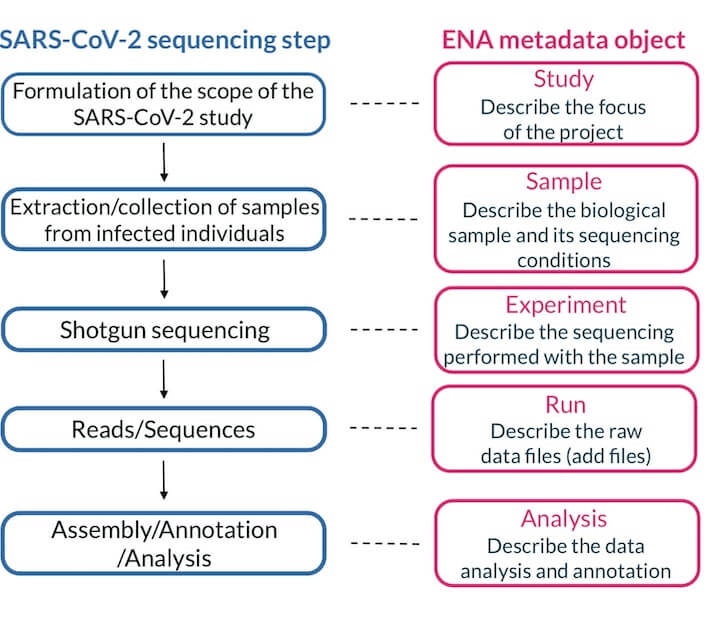
For more information on how metadata objects are used and how they are related to one another, see this video produced by ENA.
Description of sequence file formats
FASTA
A text-based file format used for nucleotide, peptide, or protein sequences. The sequences themselves are preceded by the sequence name, which comprises a single line. The sequence name is preceded by a single right-facing arrow (‘>’) that distinguishes it from the sequence itself.
A sequence in FASTA format may look e.g.:
>ENA|MT192765|MT192765.1 Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/PC00101P/2020, complete genome.
GTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCT...
AAACGAACTTTAAAATCTGTGTGGCTGTCACTCGGCTGCATGCT...
For more information on this file format, see Wikipedia on FASTA format.
FASTQ
A FASTQ file is a text-based file format that contains sequence data (including a sequence name) in a similar way as FASTA files (described above). The main difference between the two is that FASTQ files include information about data quality, whilst FASTA files do not.
FASTQ is one of the most widely used formats in sequence analysis and is often the format outputted by sequencing machines. Most analysis tools prefer FASTQ files to FASTA files because they contain more information.
The syntax used in FASTQ files is slightly different to that in FASTA files. Each sequence involves at least 4 lines:
-
Sequence Header - Comprises a sequence name and description that starts with ‘@’, instead of the ‘>’ used in FASTA files. Everything from the ‘@’ to the first whitespace character is considered to be the sequence identifier (name). Note that everything after the first whitespace is considered to be the sequence description. The sequence description follows a specific format and holds information regarding sample information.
-
Sequence - Lines of sequence data in the same format as in FASTA files (see above section describing FASTA file formats).
-
Line starting with ‘+’ - This can include the same sequence identifier and description as in the sequence header, but is typically left blank.
-
Quality Scores - The fourth line contains scores related to data quality.
For more information on this file format, see Wikipedia on FASTQ format.